
Immunotherapy, in the form of immune checkpoint inhibition (ICI), has transformed cancer care across multiple tumor types. However, existing biomarkers often fail to accurately predict which patients will draw maximal benefit. Tumor Mutation Burden (TMB) has emerged as a tumor-agnostic predictive biomarker of immunotherapy response. Measured as the number of somatic mutations per megabase (mut/Mb) of analyzed DNA sequence, TMB serves as a proxy of tumor “foreignness” to the immune system. The U.S. Food and Drug Administration (FDA) has granted approval of ICI with pembrolizumab for tumors with TMB ≥ 10mut/Mb regardless of cancer type. Despite this milestone, its clinical utility as a pan-cancer companion diagnostic for ICI is limited and TMB remains imperfect as it does not effectively and consistently predict immunotherapy response. In our recent publication (DOI), we revisit the biological, technical, and clinical nuances that limit TMB’s value and highlight how its fine-tuning might overcome these challenges.
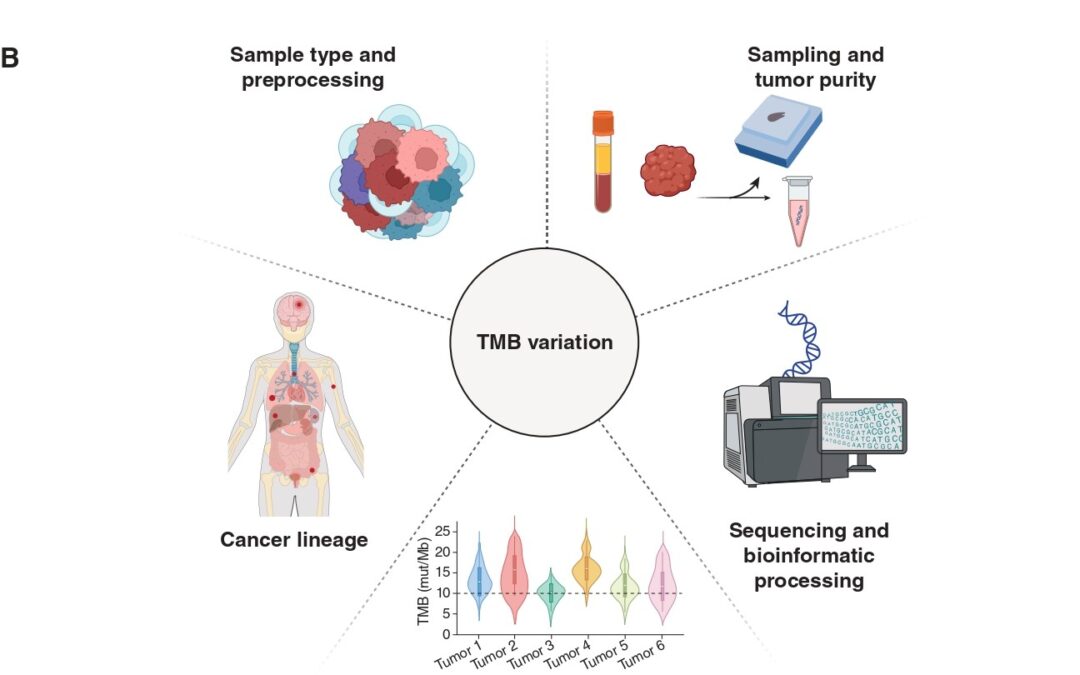
The biological premise of TMB is straightforward: more mutations = higher chances of tumor immune recognition. But this view oversimplifies a process with a high degree of complexity. Not all mutations translate into peptides, and not all peptides are immunogenic. Only a fraction of mutation-associated peptides, known as neoantigens, can be effectively presented on major histocompatibility complex (MHC) molecules on cell surface and be recognized by T-cell receptors. Hence, neoantigen quality matters more than mutation quantity. A tumor with high density of immunogenic mutation-associated neoantigens may therefore respond better to ICI than a tumor with higher TMB but poorer neoantigen quality.
TMB is a static snapshot, while cancer is a moving target. Over the course of tumor evolution, immunotherapy imposes selective pressure and bottlenecks, causing cancer clones harboring different TMB values to be eliminated or prevail through a process known as immunoediting. This Darwinian process can significantly alter tumor’s neoantigen landscape over time. Thus, TMB as a numeric or binarized value (high/low) falls short in capturing the dynamic nature of tumor evolution.
In addition, not all mutations carry the same significance when it comes to predicting immunotherapy responses. Some happen as early as the initial steps of carcinogenesis, are shared among many cancer clones and are termed as “clonal”. Others, known as “subclonal”, occur later, are shared only among few tumor cells and are more prone to loss over time. Taking a step beyond tumor architecture through a more functional scope, our group has previously shown that the genomic context of mutations plays its own role in shaping their likelihood of loss due to immunoediting. Mutations occurring in the multicopy state or found in haploid genomic regions are less likely to be eliminated. Mutations in multiple copies may be biologically protected from loss, while mutations in essential haploid genomic regions cannot be purged without compromising cell survival (DOI).
Another important consideration lies in the specimen and methodology employed to calculate TMB. Current FDA approval refers to tissue-based TMB calculation through next-generation sequencing (NGS) of a prespecified gene panel. However, this method is subject to biases related to tumor heterogeneity, sampling site, and tumor purity, especially in metastatic or heavily pretreated settings. In our piece, we touch upon the opportunities blood-based TMB estimation provides. Our paper discusses the emerging role of blood-based TMB estimation from circulating tumor-derived DNA fragments (ctDNA). Liquid biopsy offers a less invasive, real-time alternative that captures spatial and temporal heterogeneity, enabling real-time tracking of tumor mutational status throughout ICI treatment.
The approved cutoff of 10mut/Mb for ICI administration, although convenient, may not be universally applicable in the pan-cancer context. Tumor histology and lineage play a critical role in shaping mutation patterns and immune responses. For instance, malignant mesothelioma typically stands in the lower TMB spectrum, yet some patients respond to ICI. Alternative biomarkers, such as genome-wide loss of heterozygosity, may prove more useful in these contexts (DOI). Our published work includes an analysis of TMB values in correlation with ICI responses across several cancers, highlighting the discrepancies histology and lineage impose when it comes to establishing cutoffs that maximize the predictive value of TMB.
Furthermore, the overall tumor genomic landscape within which TMB is interpreted holds great significance. High TMB may result from mutagenic processes such as microsatellite instability or POLE/POLD1 mutations (ultra-mutated phenotype), which are associated with favorable ICI responses. Additionally, distinct mutational signatures, including those induced by ultraviolet light, tobacco smoke, or APOBEC enzymes, can influence immunogenicity. Finally, co-mutation patterns largely dependent on cancer lineage add an extra biological modifier of ICI response that needs to be considered when calibrating TMB through the lens of tumor biology. Example of such patterns include STK11/LKB1 mutations, which have been shown to dampen ICI response despite high TMB.
Overall, it is true that TMB is far from perfect when it comes to predicting ICI response. Looking into the future, its biological and technical standardization is expected to upgrade its predictive worth and allow for maximal leverage of its value in the clinical setting.